Protein switch in blood vessels exacerbates damage in vascular diseases
GPR153 promotes inflammatory reactions in the vessel wall in response to acute damage
After damage to blood vessels, the healing processes can spiral out of control and become pathological themselves, for example in atherosclerosis. Researchers at the Max Planck Institute for Heart and Lung Research have now shown that GPR153, a protein from the group of G protein-coupled receptors, is largely responsible for this. In the event of vascular damage, the protein acts as a switch for inflammatory activation, thereby exacerbating the damage. GPR153 could therefore be a starting point for a therapy that could help alleviate diseases of the vascular system.
G protein-coupled receptors are a large group of proteins involved in the regulation of a variety of cellular processes. They are essential for communication between cells and within cells. Although many of these receptors are known as molecules, their actual function is still unknown. One of these molecules, known as an orphan receptor, is GPR153. The protein is found in the cell membrane of both the smooth muscle cells of the vascular wall and the innermost cell layer, known as endothelial cells. Nina Wettschureck's research group at the Max Planck Institute for Heart and Lung Research has now succeeded in deciphering how the protein works.

First, the Max Planck researchers observed in mice with various vascular wall injuries that the production of the receptor in the cells was ramped up as a result of this damage. Similarly, the research team found elevated GPR153 levels in samples from patients with arteriosclerosis. ‘We interpreted this as a clear indication that GPR153 plays a role in injuries to the vascular wall,’ said Jingchen Shao, first author of the study.
After an injury, characteristic processes take place in the vessel wall: inflammatory activation occurs in the endothelial cells. This attracts more immune cells. The injury to the endothelium also affects the neighbouring smooth muscle cells. They begin to divide and migrate towards the endothelium. This can lead to the formation of a new layer in the vessel wall, known as the neointima.
‘Our study now shows that GPR 153 is an important switch for these processes, but in a negative sense,’ says Shao.
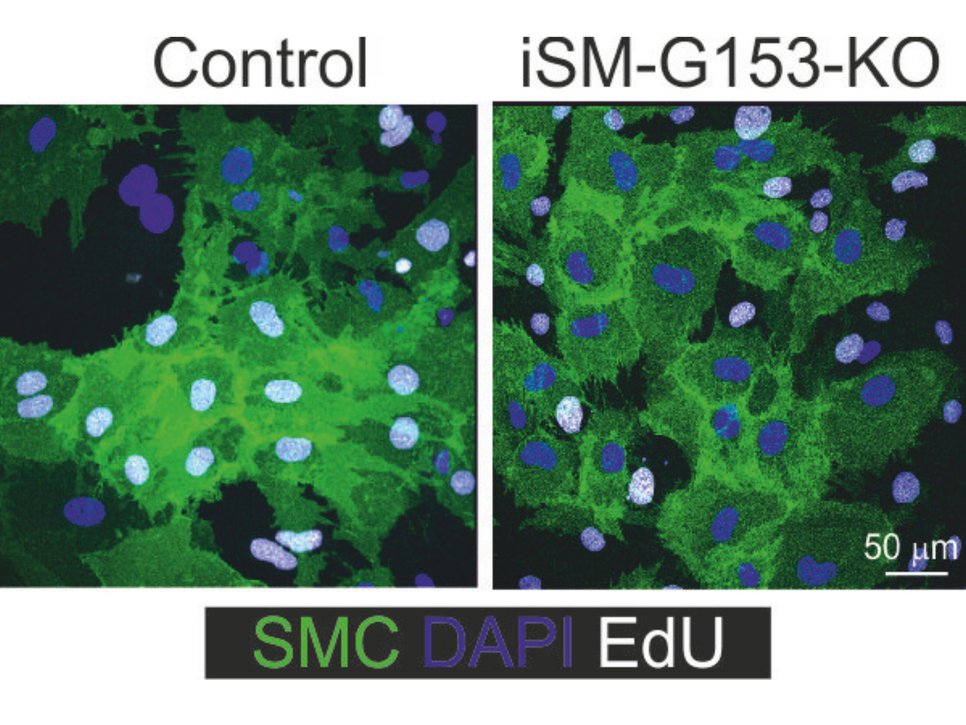
In mice lacking GPR153 in smooth muscle cells, the growth of smooth muscle cells after damage is significantly reduced and neointima formation is correspondingly diminished.
‘Conversely, this means that GPR153 promotes and intensifies damage to the vessel wall,’ says Shao.
, they divide significantly less than when GPR 153 is present. This can be seen in the white-coloured cell nuclei. The smooth muscle cells are coloured green. Cell nuclei that have not divided are shown in blue.")
In further experiments, the Bad Nauheim researchers showed how GPR153 works at the molecular level: The focus is on a messenger substance in the cells called cAMP. GPR153 slows down its production; if GPR153 is missing, a significantly higher cAMP level develops. This has consequences for the cells and the damage to the vessel wall: "On the one hand, signalling pathways that tend to protect the cells become more active. On the other hand, high cAMP levels slow down the signalling pathways that intensify inflammatory reactions and stimulate cell division," explains Nina Wettschureck, group leader at the Max Planck Institute and head of the study.
Interestingly, very similar effects are observed when GPR153 is switched off in endothelial cells: due to reduced inflammatory activation, fewer immune cells adhere to the endothelium and migrate into the tissue. This dampens the inflammatory response and thus limits the damage. Mice in which GPR153 was specifically switched off in the endothelial cells of the brain showed impressive improvements in models of multiple sclerosis and stroke compared to control animals: ‘We found fewer inflammatory foci in the brains of these mice, fewer immune cells had migrated, and the animals were neurologically less conspicuous,’ Shao summarises the data.
From the perspective of the Max Planck researchers, the findings of the study point to new therapeutic targets.
‘Our data show that GPR153 amplifies damaging processes in injured blood vessels. Therefore, the next step is to investigate ways to specifically inhibit its production in vascular damage or block the activity of the protein,’ says Wettschureck.
 are enormously successful drug targets, but the function of some GPCR is still completely unknown. One of these enigmatic receptors is GPR153. GPR153 is upregulated in vascular cells in response to damage and contributes to their inflammatory activation and dedifferentiation.Cell-type specific inactivation of GPR153 (indicated in green) prevents smooth muscle cell dedifferentiation and endothelial inflammation. As a consequence, cell type-specific knockout mice are protected in disease models such as neointima formation, stroke or multiple sclerosis.In summary, GPR153 is an interesting target for parallel inhibition of endothelial inflammation and smooth muscle dedifferentiation.")
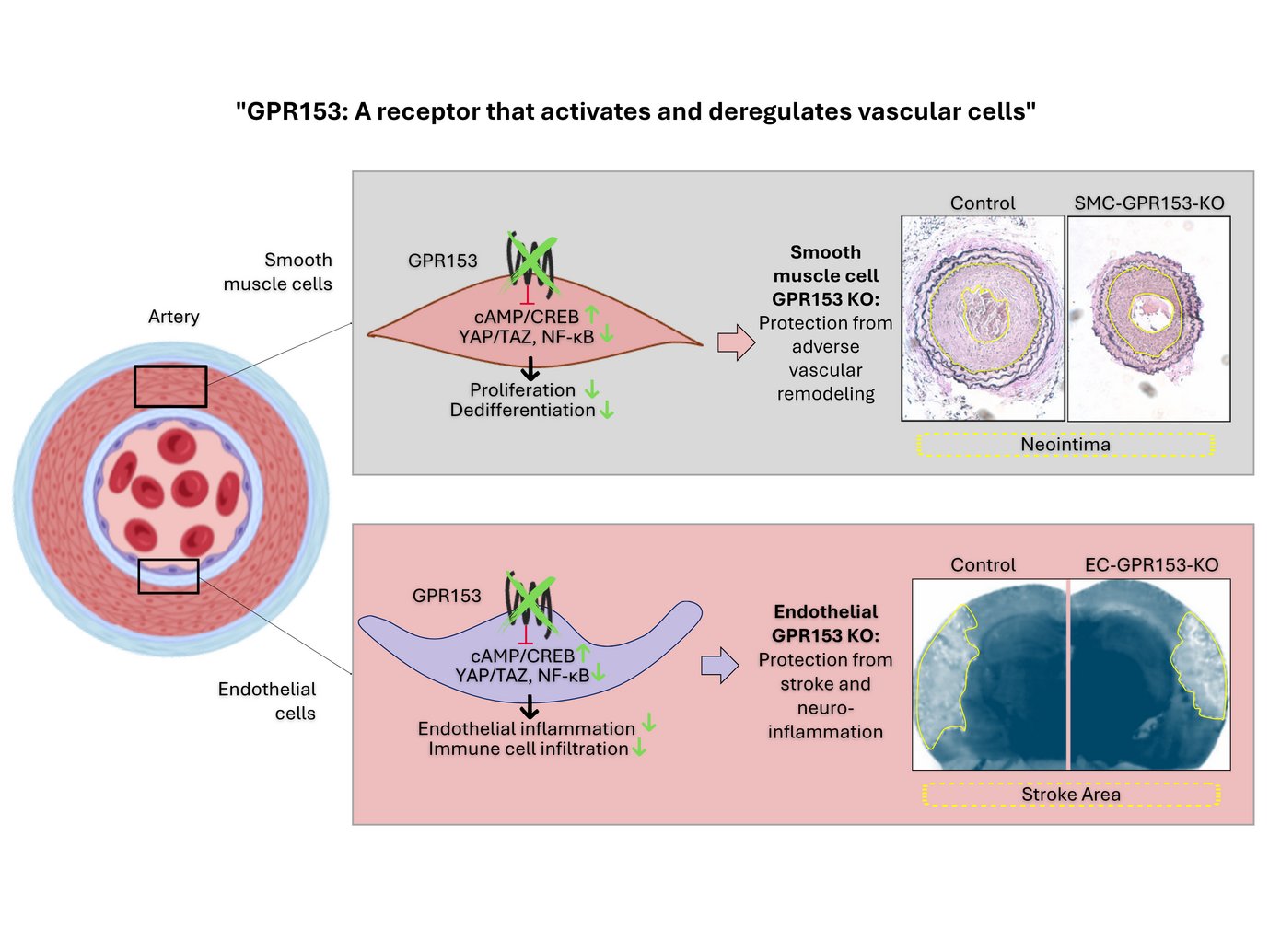
Cell-type specific inactivation of GPR153 (indicated in green) prevents smooth muscle cell dedifferentiation and endothelial inflammation. As a consequence, cell type-specific knockout mice are protected in disease models such as neointima formation, stroke or multiple sclerosis.
In summary, GPR153 is an interesting target for parallel inhibition of endothelial inflammation and smooth muscle dedifferentiation.